 Fraunhofer Institute for Mechanics of Materials IWM
Fraunhofer Institute for Mechanics of Materials IWMSolar cell research from theroretical calculations
Hybrid organic-inorganic halide perovskites are the most promising photovoltaic absorber materials to substitute or complement silicon for high-efficiency solar cells. These hybrid materials are often constrained by their low stability and critical elements like lead. Computational high-throughput-screening studies, based on solid-state electronic-structure theory, are useful to identify promising substitute materials with targeted properties. However, an accurate description of the electronic structure at device operating temperatures is highly challenging and, thus, the predictive power of previous computational studies has been limited. Consequently, ideal lead-free absorber materials have still not been identified and efficiencies of lead-based materials hold further optimized potential.
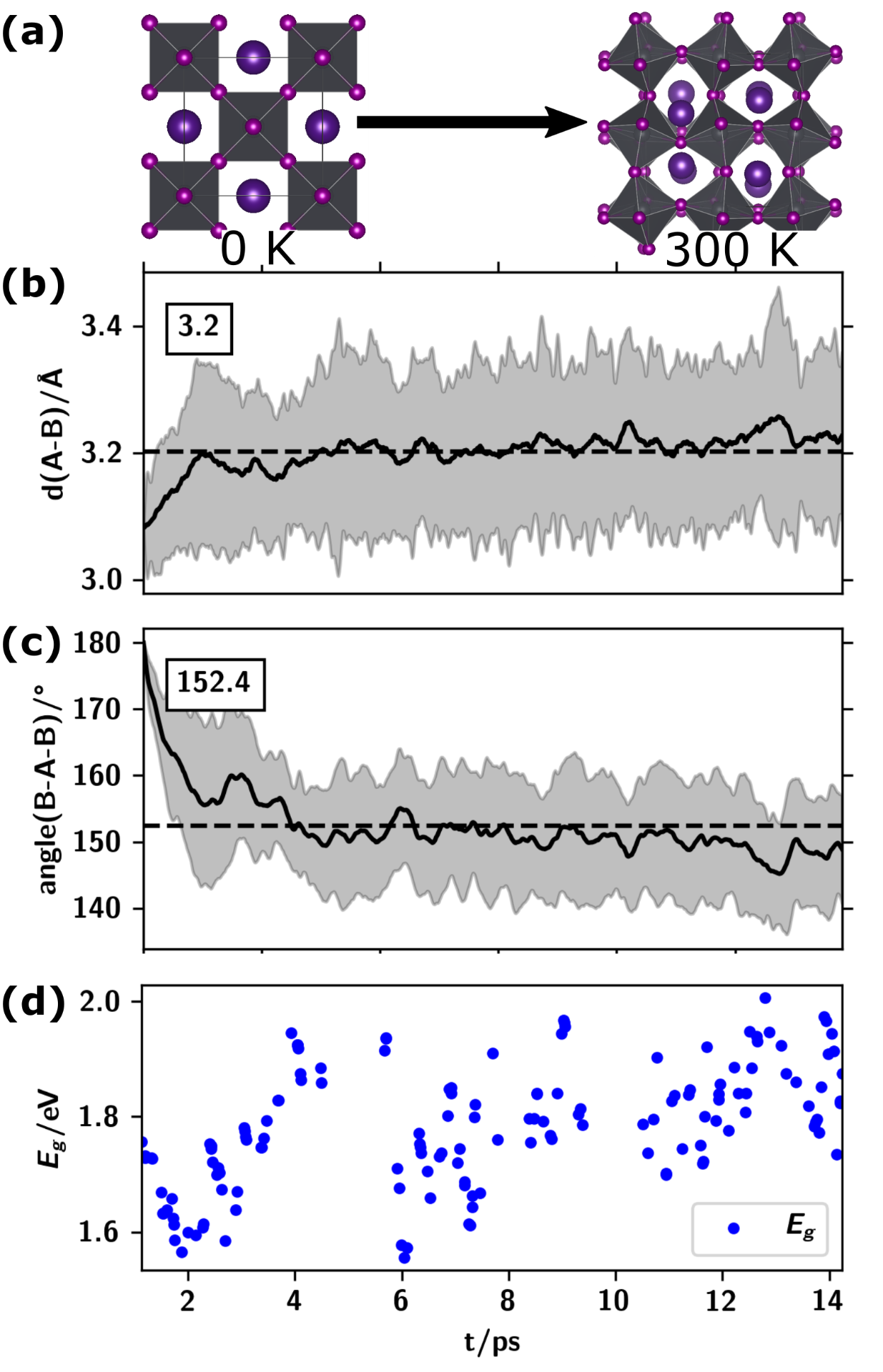
Despite being observed as time and spatial average, the structure of perovskites at finite temperature is not the ordered cubic phase, but a locally distorted and tilted structure. This is observed in ab initio molecular dynamics calculations, monitoring bond distances (b) and angles (c) of the perovskite BX6 octahedrons. These structural changes have a severe influence on the electronic structure, raising the observed electronic band gap of, e.g., CsPbI3 from ~1.0 eV to ~1.78 eV.
In the Fraunhofer Lighthouse project MaNiTu we tackle these aspects. On one hand, we aim to identify new absorber materials that contain neither lead nor any other critical element. We use a combination of automated literature screening of harvested literature data and high-throughput first-principles calculations. On the other hand, reference compounds that currently provide best power conversion efficiencies need to be understood on a better qualitative as well as quantitative level. In particular, the dynamical structure of the absorber materials drastically influences the optical and electronic properties, leading to significant discrepancies between standard calculations (employing models at zero temperature) and experimental measurements (obtained around room temperature). We developed an efficient computational workflow, based on density-functional theory, which is suitable to predict band gaps for arbitrary compounds reliably and in good quantitative agreement with experimental band-gap data. This allows to analyze intensively studied absorber materials such as (HC(NH2)2)xCs1-xPb(IyBr1-y)3 (where x and y may be varied between zero and one) in detail, to predict the properties of novel compounds with better accuracy, and to further analyze the interface properties.
Fraunhofer Lead Project »MaNiTu«
Networks within the Fraunhofer-Gesellschaft - Fraunhofer IWM
Back to Assessment of Materials and Lifetime Concepts Highlights